
Primer: Genome Sequencing

Overview
Genome sequencing refers to the process of determining the exact order of the entirety of nucleotides in an organism’s genome at a single time. Currently, genome sequencing has contributed to identifying inherited disorders, monitoring global disease outbreaks, establishing mutation frequencies in cancer, and revealing additional avenues of research in genomics. Although sequencing technologies have become increasingly accessible and affordable in recent years, scientists have yet to establish an efficient and accurate method to map short reads. As a result, the majority of repetitive or highly variable regions within the genome remain difficult to assemble.
In this module, we will outline the primary methods of sequence analysis and their known applications, and compare the advantages and disadvantages of various genome sequencing technologies.
Sequencing, Re-sequencing, & Genotyping
Sequencing is an approach that allows scientists to determine the exact order of nucleotides in DNA or RNA, or the order of amino acids in proteins. Currently, scientists primarily rely on whole-genome sequencing for clinical applications, including the unbiased diagnosis of rare diseases, exploration of antimicrobial resistance, and the assessment of regulatory variation.
Re-sequencing is an approach that characterizes a sample genome and its associated variation by mapping and aligning short, region-specific sequence reads to an existing reference genome. Compared to sequencing, re-sequencing relies on a reference sequence to facilitate genome assembly and alignment. Currently, re-sequencing is primarily used to identify variable regions in human genomes.
Genotyping is an approach that detects genetic markers that contribute to major phenotypic changes by comparing a DNA sequence to that of another sample or reference sequence. Although genotyping only targets known markers within a sequence of interest, this is often sufficient for the majority of use cases, and is also cheaper to execute compared to sequencing. Currently, genotyping is used to identify single nucleotide variants, copy number variants, and structural changes in DNA.

In short, sequencing accounts for every single nucleotide, re-sequencing considers certain regions of interest, and genotyping only targets specific biomarkers of interest.
Genome Sequencing Technologies
Sanger Sequencing
Sanger sequencing, developed by Frederick Sanger and his colleagues in 1977, is an approach to DNA sequencing that selectively incorporates chain-terminating dideoxyribonucleotides (ddNTPs) by DNA polymerase during DNA replication. Sanger sequencing was primarily used in the Human Genome Project to sequence small fragments of human DNA, which were then used to construct larger fragments and eventually, assemble entire chromosomes.
Compared to other sequencing technologies, Sanger sequencing is cost-effective for smaller fragments of DNA, and is comparatively lower-throughput. However, Sanger sequencing is characteristic of low sensitivity and scalability, and can be expensive for larger regions of DNA.

Sanger Sequencing Workflow
Library Preparation: Genomic DNA is used as a template for chain-terminating PCR. To distinguish between various nucleotides, ddNTPs are tagged with a different fluorescent label, and are mixed within the same reaction.
Sequencing: Oligonucleotides are run through gel electrophoresis within the sequencing machine.
Data Analysis: A computer reads each band of the gel sequentially, and relies on fluorescence to determine the identity of each terminal ddNTP. Since each of the four ddNTPs is tagged with a different fluorescent label, the fluorescent peak of each nucleotide along the length of the template DNA will reveal the resulting sequence.
Next-Generation Sequencing (NGS)
Next-generation sequencing relies on massively parallel sequencing to produce short reads, enabling the interrogation of hundreds to thousands of genes across multiple samples. Compared to Sanger sequencing, NGS is more cost-effective, accurate, and reliable, and is capable of sequencing an entire genome at once. Furthermore, NGS provides more comprehensive genomic coverage, higher analytical sensitivity, and greater resolution of genomic variants. However, compared to other sequencing technologies, NGS is more expensive when analyzing a low number of samples, and the process of amplification is time-consuming, often resulting in errors or bias in NGS output.
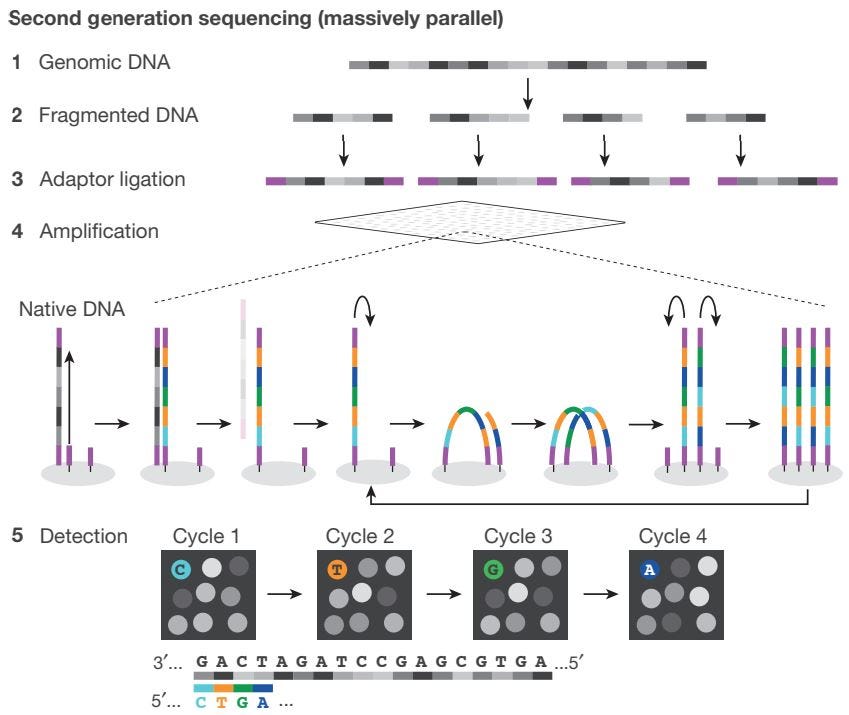
NGS Workflow
Library Preparation: Genomic DNA is split into smaller fragments. Adaptors containing complementary sequences are added to the ends of the DNA fragments, to facilitate binding to the flow cell. Fragments are then amplified and purified.
Sequencing: Libraries are loaded onto a flow cell and placed on the sequencer. Clusters of DNA are amplified, resulting in millions of copies of single-stranded DNA.
Data Analysis: Instrument software performs base calling to identify the resulting nucleotide sequence.
Example: Illumina Sequencing-by-Synthesis (SBS)
Illumina Sequencing-by-Synthesis (SBS) is an approach for next-generation sequencing that relies on a reversible termination method to detect individual nucleotides as they are incorporated into elongating DNA strands.
Illumina Sequencing-by-Synthesis (SBS) Workflow
Sample Preparation: Adaptors are added to the ends of the DNA fragments, and additional motifs (i.e. sequencing binding sites, indices, and regions complimentary to flow cell oligonucleotides) are introduced through reduced cycle amplification.
Cluster Generation: Polymerase creates a complement of the hybridized fragment on the flow cell, and strands are clonally amplified through bridge amplification.
Sequencing: 3’ ends are blocked to prevent undesirable priming, and the first sequencing primer is extended to produce the first read. The total number of cycles determines the final read length.
Data Analysis: For each sample, reads with similar stretches of base calls are locally clustered. Forward and reverse reads are paired to create contiguous sequences, which are aligned to the reference genome for variant identification.
Single-Molecule, Real-Time (Long-Read) Sequencing
Single-molecule, real-time sequencing relies on patterns detected from ion flow to establish genome sequences. As nucleotides are passed through a protein nanopore, changes to an electrical current are monitored to determine the genome sequence. Currently, long-read sequencing is used to sequence highly repetitive genome sequences and generate long reads for de novo genome assembly.
Compared to other methods, long-read sequencing allows for rapid access to time-critical information, such as pathogen identification, as well as the ability to terminate sequencing once target results have been obtained, allowing for washing and reuse of the flow cell. However, long-read sequencing is relatively low-throughput compared to NGS, and is also characteristic of high error rates randomly distributed across sequences.
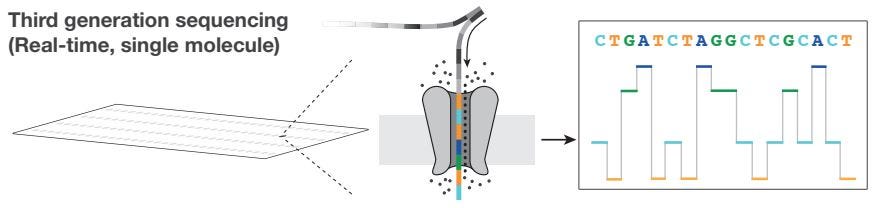
Long-Read Sequencing Workflow
Sample Preparation: DNA/RNA, motor protein, and adaptor sequence are inserted into a flow cell. Flow cells contain multiple membrane wells, each with a single nanopore. The tether attaches to one strand of the sequence, while the other strand is inserted into the nanopore.
Sequencing: Nucleotide base pairs disrupt the ionic current, which is measured in single trace.
Data Analysis: Patterns within the electrical current are interpreted to reveal the resulting genome sequence.
Example: Oxford Nanopore Sequencing
Oxford Nanopore Sequencing is an approach for long-read sequencing that relies on flow cells containing nanopores embedded in an electro-resistant membrane. Within the flow cell, each nanopore corresponds to a unique electrode that measures the electrical current flowing through the nanopore. When the current is disrupted, the resulting squiggle is decoded using base calling algorithms to provide a real-time evaluation of the input genome sequence.
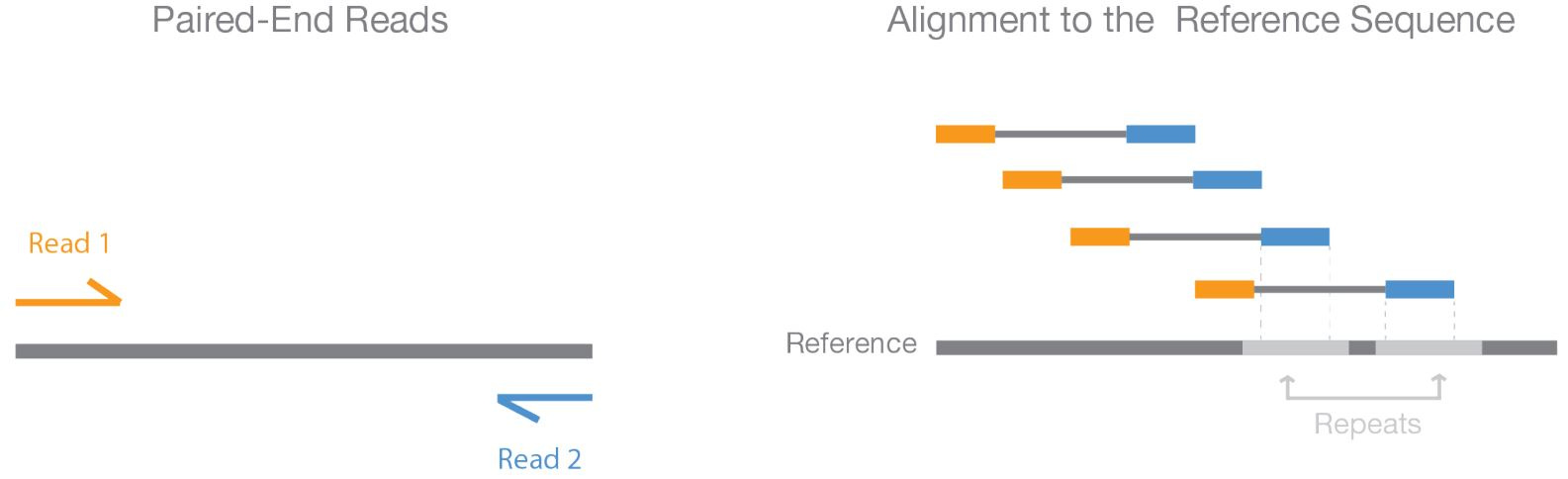
Paired-End Sequencing
Paired-end sequencing is an approach that sequences both ends of a DNA fragment of an approximately known length, effectively linking the end sequences. Since the distance between primers is known, fragment length is established before sequencing begins, allowing for more straightforward genome assembly. Currently, paired-end sequencing provides high-quality alignment across repetitive regions, and is primarily used to detect common DNA rearrangements, such as insertions, deletions, and inversions. However, paired-end sequencing tends to be more expensive compared to other sequencing technologies, as it requires an additional round of sequencing and supplemental reagents.
References
[1] Shendure, J., Balasubramanian, S., Church, G. et al. DNA sequencing at 40: past, present and future. Nature 550, 345–353 (2017). https://doi.org/10.1038/nature24286
[2] Next-Generation Sequencing (NGS) | Explore the Technology. www.illumina.com/science/technology/next-generation-sequencing.html
